Abbreviated New Drug Application (ANDA) and Abbreviated New Drug Submission (ANDS)
Generic drugs are a low-cost, safe, and effective alternative to brand name drugs for consumers across the world. Similar to brand name drugs, before a generic drug can be sold in US or Canada Markets it must be approved by FDA in US or by PDD in Canada. To obtain such approvals, companies must submit an Abbreviated New Drug Application (ANDA) in US or an Abbreviated New Drug Submission (ANDA) in Canada. These applications don’t require preclinical and clinical data to demonstrate the safety and efficacy of the drug but focus instead on firmly establishing the bioequivalence between the generic and innovator (brand name) drug.
Our Services:
We provide comprehensive writing and regulatory support for IND submissions including:
Regulatory consulting and development strategy
Data readiness review and data analytics support
Non-clinical content review
Written and tabulated summaries of non-clinical data (pharmacology, toxicology, etc.)
Clinical study Reports (when prior clinical data is available)
Investigator’s Brochures (IBs)
Clinical protocols
Informed Consent Forms
Pre-IND meeting participation and support
Our regulatory team will work with you to collect the necessary information, prepare the submission filings, and assist with meetings and discussions with the US and Canada regulatory bodies towards a successful outcome of your pharmaceutical application.
DRUG REGULATORY CONSULTING
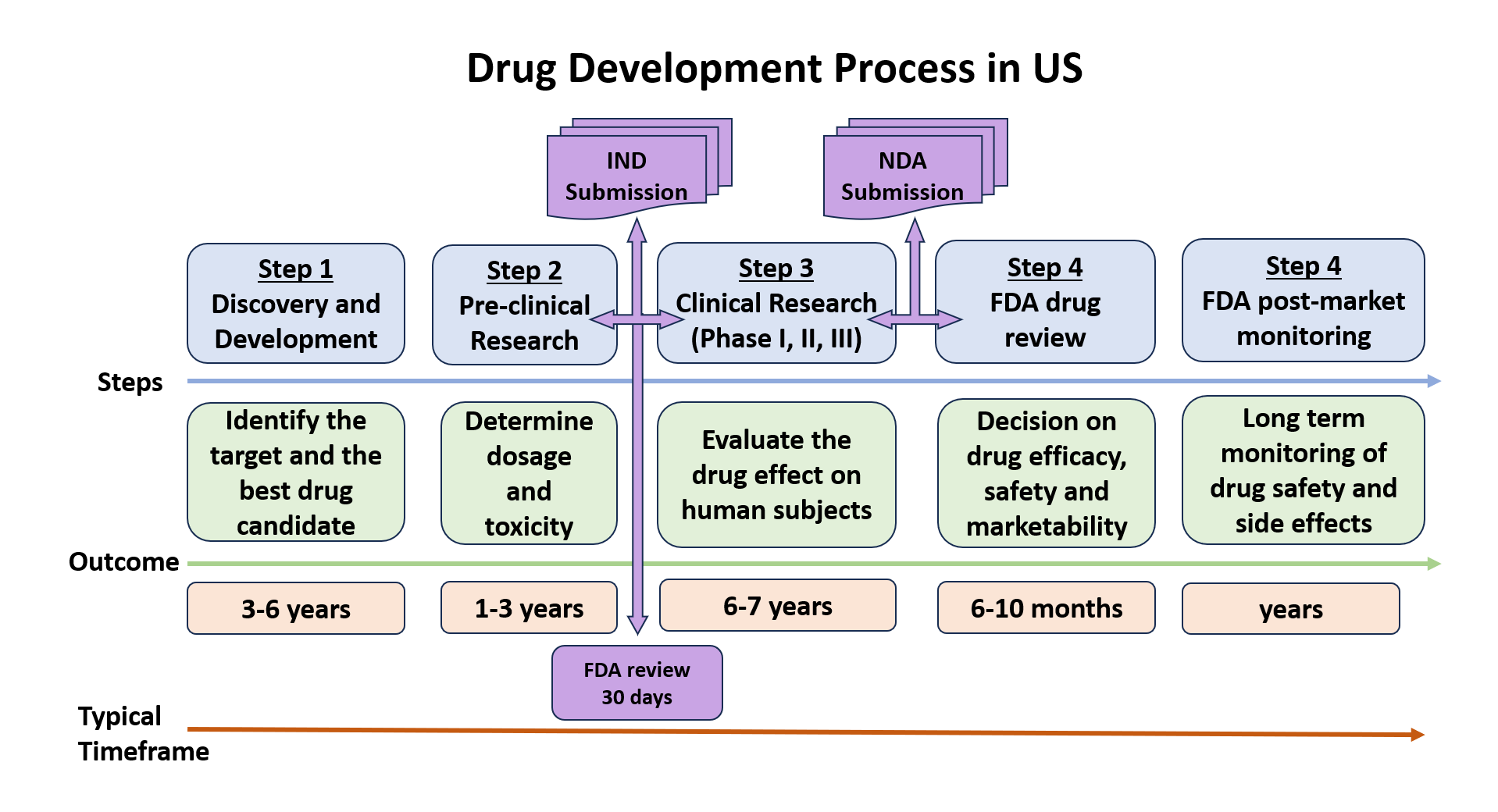
IND (Investigational New Drug) Application
US Federal law requires any drug that needs to be transported and distributed across state lines to have an approved marketing application. A sponsor interested in shipping an investigational drug to clinical sites in multiple US states must obtain an exemption to this legal requirement in the form of an approved IND application.
There are two IND categories: Commercial and Research (on-commercial), and three IND types: An Investigator IND, Emergency Use IND and Treatment IND (see 21CFR Part 312 and FDA/IND Application for more details).
The IND application must be submitted in eCTD format and contain documentation structured in five modules: Administrative and Prescribing Information, Summaries, CMC (Quality) Information, Nonclinical Study Reports and Clinical Study Reports. A Drug Master File (DMF) might be required to accompany the submission.
The application is reviewed by FDA-CDER to ensure that trial participants will not be subject to unreasonable risks. Trials may begin if no clinical hold is issued by FDA in 30 calendar days from submission.
A typical timeline to prepare (write) an IND application is 3-4 months (~180 documents, ~1500 pages). Participation in a pre-IND meeting with the FDA authority and presenting a complete documentation and compelling safety data is highly recommended.
New Drug Application (NDA) and New Drug Submission (NDS)
Companies interested in launching a new pharmaceutical drug in US or Canada must first submit a New Drug Application (to the FDA in US) or a New Drug Submission (NDS) (to the Health Canada’s Pharmaceutical Drug Directorate (PDD)). These regulatory filings are meant to confirm and substantiate the pharmaceutical safety and efficacy of the drug before it reaches the market.
NDA and NDS submissions enclose a comprehensive overview of the drug including important information about its active ingredients, its pharmacokinetics and pharmacodynamics properties as well as details about how the drug is manufactured, processed, packaged, and labelled.

